RetroExplainer
论文标题:Retrosynthesis prediction with an interpretable deep-learning framework based on molecular assembly tasks
期刊:Nature Communications(2023)
Abstract
1.现有深度学习逆合成方法的问题
- 大多数深度学习方法都难以解释,如同一个“黑箱”,其中蕴含的insight较少。
2.本文工作
- 提出RetroExplainer,将逆合成任务形式化为分子组装过程
- 分子组装过程包含多个由深度学习引导的逆合成操作步骤
3.模型的核心部件
- 多感知、多尺度图Transformer
- 具有结构感知能力的对比学习
- 动态自适应多任务学习
4.单步逆合成实验结果
- 在12个大规模benchmark数据集上的结果证明了RetroExplainer的有效性,其表现优于现有的SOTA单步逆合成方法
5.分子组装的可解释性
- 赋予模型良好的可解释性,使得决策过程透明化,允许进行定量归因。
6.多步逆合成实验结果
- 成功识别出101条路径,其中86.9%的单步反应是已有文献报道过的。
Introduction
1.分子逆合成任务
- 为目标分子的高效合成,确定一组合适的反应物。
2.基于模板的逆合成方法
- 依赖于反应模板将产物转化为反应物
- 通常使用分子指纹,结合
MLP,以编码分子产物,并推荐合理的模板。 - 举例
Segler等人利用扩展连接性指纹(Extended-Connectivity Fingerprints, ECFPs),结合扩张策略网络来指导模板搜索。Chen等人采用一种类似于单步逆合成预测器的策略,用于神经网络引导下的多步逆合成规划。
- 问题
- 构建反应模板的过程,目前仍然依赖于人工编码或者复杂的子图同构算法,使得在庞大的化学空间中探索潜在的反应模板变得困难。
3.半模板、无模板逆合成方法
- 目的:解决基于模板方法难以探索潜在模板的问题。
- 分子表示:利用分子指纹技术获取分子级别的表示。
- 举例
Chen等人引入FeedForward EBM (FF-EBM)方法,由无模板模型进行补充与完善(complemented by template-free models)。
- 分类
- 基于序列的方法:将分子表示为线性化的字符串。
- 基于图的方法:将分子表示为图结构。
4.基于序列的逆合成方法
- 产物分子的表征:使用线性化记号,如
SMILES(simplified molecular-input line-entry system)。 - 举例
Liu等人引入了Seq2Seq模型,该模型包含双向长短时记忆(LSTM)编码器和解码器以进行逆合成转换。Karpov等人针对逆合成分析调整了Transformer架构的学习率调度策略,并采用快照学习方法。Tetko等人提出一种基于Transformer的逆合成模型,通过SMILES增强提高了性能。- 随着预训练-微调范式的兴起,
Irwin等人提出了MolBART,利用大规模自监督预训练,加速逆合成任务的收敛。
- 方法局限性
- 线性化的分子表示方式(如SMILES)难以直接探索结构信息和原子性质,这些内容对于逆合成分析至关重要。
- 基于
SMILES的分子表示方法语法严格,语义有效性不足,容易导致频繁出现无效语法。
- 解决方法
- 其他一些方法被提出,以避免无效字符串的生成,或嵌入更为丰富的结构信息。
5.基于图的逆合成方法
- 两阶段范式实现
- 反应中心预测(
RCP, Reaction Center Prediction) - 合成子完成(
SC, Synthon Complesion)。
- 反应中心预测(
- 应用——正向反应预测
Jin等人将其应用于正向反应预测,提出使用Weisfeiler-Lehman同构测试[30]图学习来预测反应结果。
- 基于
GNN框架的模型发展- 随着
GNN的发展,许多基于GNN的框架出现于逆合成领域,并且在性能上取得了显著的提升。 Shi等人提出G2G框架,利用关系图卷积网络(R-GCN)进行RCP,并结合强化学习进行SC。Yan等人提出的RetroXpert应用了图注意力网络(GAT)的变种进行RCP,以及基于序列的Transformer进行SC。Somnath等人提出的GraphRetro为两个阶段设计了两种消息传递神经网络(MPNNs)。Dai等人提出GLN方法,利用反应模板连接产物和反应物。
- 随着
- 传统基于
GNN方法的问题- 主要关注分子的局部结构,忽略了长距离特征(如范德华力)的影响。
- 问题解决
Ying等人提出Graphormer,引入了基于最短路径的方法进行多尺度拓扑编码。
- 直接模拟图变化的方法以外的其余方法
- 其他基于图的方法通过翻译反应物,以预测图变化。
6.现有逆合成方法的问题
- 信息表征的不足
- 基于序列的方法:处理分子时,丧失先验信息。
- 基于图的方法:忽视顺序信息、长距离特征。
- 可解释性差
- 现有模型的决策机制尚不清晰,极大限制模型可靠性与实际应用价值。
- 无法解释模型的工作原理,或提供实质性的
insight。
- 单步逆合成、反应物不易获取
- 大多数现有方法侧重于单步逆合成预测,能够生成看似合理,但可能不易购得的反应物,
- 通常伴随手工筛选预测结果的繁琐过程。
- 从产物到容易获得的反应物,进行多步逆合成预测和路径规划,更有实际意义。
7.论文贡献
-
提出
RetroExplainer——一种结合化学知识、深度学习引导的分子组装方法,用于进行具有量化可解释性的逆合成预测。 -
贡献一——构建强大、信息丰富的分子表征
- 设计一种多感知、多尺度图
Transformer(Multi-Sense Multi-Scale Graph Transformer, MSMS-GT),用于通用的分子表征学习。 - 使用动态适应性多任务学习(
Dynamic Adaptive Multi-Task Learning, DAMT),以实现平衡的多目标优化。 - 使用结构感知对比学习(
Structure-Aware Contrastive Learning, SACL),以捕捉分子结构信息。 - 结果:
RetroExplainer在包括三个常用数据集(USPTO-50K、USPTO-FULL和USPTO-MIT)在内的共12个大规模benchmark数据集上表现优秀。
- 设计一种多感知、多尺度图
- 贡献二——良好的可解释性
- 引入分子组装过程
- 提供透明的决策过程。
- 生成能量决策曲线,将预测分解为多个阶段,并允许在子结构级别进行归因分析。
- 有助于理解“反事实”预测,揭示数据集中潜在的偏置。
- 引入可解释逆合成预测
- 提供更细致的参考信息(例如:对某一特定化学键断裂的置信度),启发研究人员设计定制化的反应物。
- 引入分子组装过程
- 贡献三——确保预测产物的可合成性、避免手动筛选候选反应物
- 做法:整合
RetroExplainer模型与Retro算法。 - 结果:对
101种复杂药物分子进行合成路线,86.9%的单步反应都能在文献报道中找到对应。
- 做法:整合
Results
一、基于USPTO数据集的性能比较
1.实验设置
- 三个常用
USPTO的benchmark数据集:USPTO-50K, USPTO-FULL, USPTO-MIT。 - 比较:与
21种逆合成方法进行对比。 - 数据集分割方法:与以往工作相同的随机分割方法。
2.USPTO50K数据集实验结果(表1)
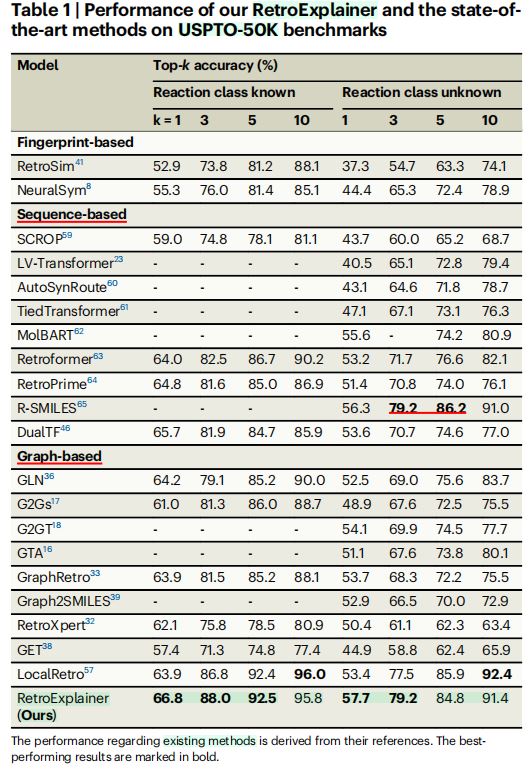
- 性能评估标准:
top-k精确匹配准确度,k分别设置为1, 3, 5, 10。 RetroExplainer实验结果- 在
5项指标上,取得最优水平。k = 1, 3, 5时,针对已知反应类型。k = 1, 3时,针对未知反应类型。
k = 10的实验结果- 未达到最优精确度
- 在已知/未知反应类型的条件下,精确度接近最优模型
LocalRetro,与其差距分别为0.2%和1%。
- 综合结果(不同
top-k下的平均精度)- 分别根据是否提供反应类别信息,综合考虑
top-1, top-3, top-5, top-10预测的平均精度,RetroExplainer均获得最高精确度。 - 相比于第二名模型(已知反应类型为
LocalRetro,未知反应类型为R-SMILES),平均精度分别改进1%和0.1%。
- 分别根据是否提供反应类别信息,综合考虑
- 在
3.现有数据分割方法的问题及解决方案
- 骨架评估偏差与信息泄露
- 在随机分割的数据集中,相似分子可能同时出现于训练集和测试集中,容易导致训练数据的信息泄露,进而使模型评估产生偏差。
- 解决方案
- 采用
Kovács等人提出的基于Tanimoto相似性的数据分割方法。 - 考虑
3种相似度阈值(0.4, 0.5, 0.6)和3种测试集分割比例(0.2, 0.25, 0.3),由此产生了九种基于Tanimoto相似性的数据集。 - 在上述
9个数据集上,将RetroExplainer与R-SMILES和LocalRetro两种现有的最优方法进行比较。
- 采用
- 结果(图
2)
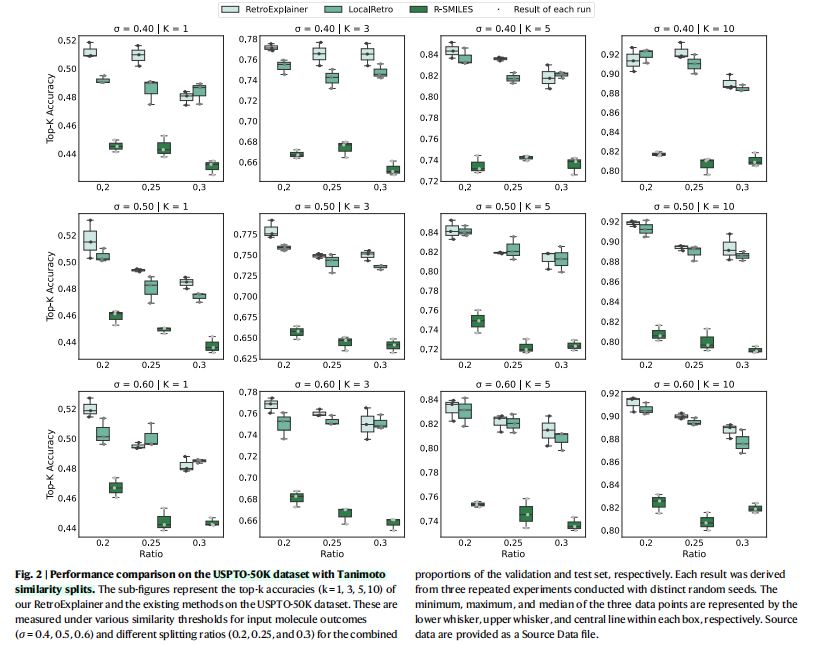
- 针对上述
9个数据集中的大部分数据集,RetroExplainer在top-1, top-3, top-5, top-10精确匹配准确度上,均超过baseline方法。
- 针对上述
- 结论
- 进一步证明了
RetroExplainer的有效性、健壮性。 - 相比现有方法,
RetroExplainer在处理未曾见过的分子骨架结构时,能展现出更强的领域适应性。
- 进一步证明了
4.大型数据集USPTO-Full和USPTO-MIT的实验结果
- 结果(表
2)
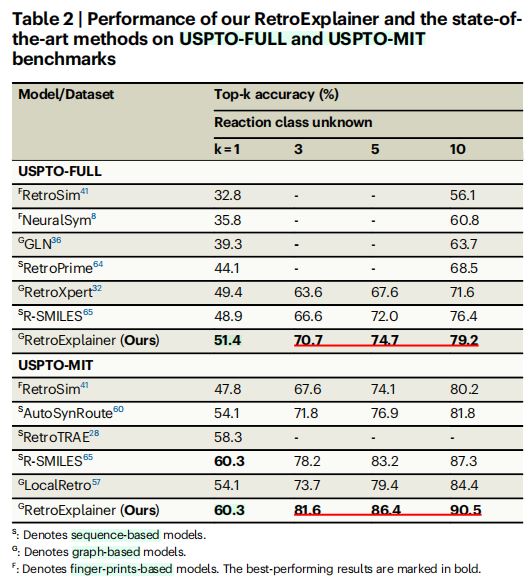
- 对于两个数据集的
4中评价指标(top-1, top-3, top-5, top-10精确度)中,RetroExplainer均以较大优势取得最佳性能。 USPTO-FULL数据集:RetroExplainer在top-1, top-3, top-5, top-10精确度上,分别比第二名的R-SMILES高出2.5%, 4.1%, 2.7%, 2.8%。USPTO-MIT数据集:结果类似。
- 对于两个数据集的
- 结论
- 在更大规模数据集的评估条件下,
RetroExplainer与以往的最先进相比方法,更为有效和稳定。 RetroExplainer更适合大规模训练场景的应用,具有更大潜力。
- 在更大规模数据集的评估条件下,
- 补充研究(后文)
- 消融研究:讨论尺度信息、增强策略模块的有效性。
- 案例研究:说明
MSMS-GT如何聚焦于分子的多尺度结构。
5.模型SOTA性能的解释
RetroExplainer取得更有表现的可能原因:数据建模的独特方法。- 两种分布的同时捕捉
- 提供产物分子图时,反应中心(
RC, Reaction Centers)和离去基团(LG, Leaving Groups)之间的联合条件分布。- 通过联合分布,考虑离去集团对反应中心的影响,使得模型可以学习信息更丰富的表示。
- 在确定反应中心、离去集团和产物后,离去集团之间连接关系的条件分布。
- 提供产物分子图时,反应中心(
- 效果
- 增强模型泛化能力(尤其是涉及多个反应中心和离去集团的情形)
- 避免了由于多个离去集团(或反应中心)问题,而产生的过度迭代需求。
- 通过学习合成子连接关系的条件分布,
RetroExplainer能够有效适应更庞大、更复杂的数据集。
二、RetroExplainer模型架构及其可解释的insight
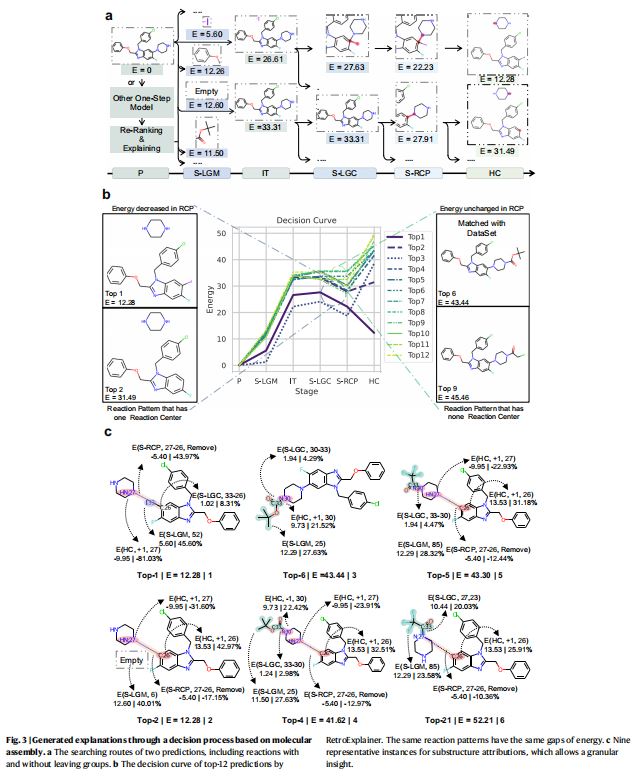
1.深度学习引导的六阶段透明决策过程(图3a)
- 原始产物(
P, Product) - 离去基团匹配(
S-LGM, Leaving Group Matching) - 初始化(
IT, Initializing) - 离去基团连接(
S-LGC, Leaving Group Connecting) - 反应中心键变化(
S-RCP, Reaction Center Bond Changing) - 氢原子数量变化(
HC, Hydrogen Number Changing)
2.决策阶段的“能量分数(energy score)”
- 各阶段依据对最终决策的贡献,生成相应的能量分数。
- 能量分数来源:由经过训练的模块计算得出。
- 离去基团匹配模块(
LGM, Leaving Group Matching) - 带氢原子的反应中心预测模块(
RCP-H, Reaction Center Prediction for Attached Hydrogens) - 键反应中心预测模块(
RCP-B, Reaction Center Prediction for Bond) - 离去基团连接模块(
LGC, Leaving Group Connecting)
- 离去基团匹配模块(
- 能量分数计算细节:见补充信息笔记
4。
3.六阶段决策过程的具体描述
- 从
P阶段出发,将该阶段的能量分数初始化为0。(在图3a中记为\(E\)) - 在
S-LGM阶段,根据LGM模块给出的概率预测,选择一个离去基团(LG),并基于LGM的概率预测,为该离去基团赋予一个能量分数。 - 计算
IT阶段的能量分数。该步骤通过添加S-LGM阶段所选离去基团的能量,以及由RCP-H、RCP-B和LGM模块各自预测出的相应事件的概率而完成。 - 在
LGC和RCP阶段,使用动态规划算法扩展搜索树中的所有可能结点。选择了概率大于设定阈值的事件,并据此给定各阶段的能量分数。 - 针对每个原子,调整氢原子数量和形式电荷,确保修改后的分子图遵循价键规则;最后,根据氢原子数量变化(
HC, Hydrogen Number Changing)的成本,计算出最终的能量分数。
4.模型决策过程的作用与解释
RetroExplainer生成的决策曲线的作用- 揭示模型为何可能会做出“错误”(化学上合理,但与数据集中记录的实际答案不符)的预测
- 有助于识别训练数据集中的潜在偏差(
bias)。
- 举例(图
3b)- 现象一
- 通过胺脱保护法合成目标产物的“正确”答案在模型中排名第
6,排名第1的预测则是C-N偶联反应。
- 通过胺脱保护法合成目标产物的“正确”答案在模型中排名第
- 解释
- 经研究发现,氢原子数量变化(
HC, Hydrogen Number Changing)阶段是导致能量分数差异的关键阶段。 RetroExplainer在HC阶段,倾向于预测胺基氢原子数量增加的反应物,表明模型在类似的分子骨架中存在HC偏向性。
- 经研究发现,氢原子数量变化(
- 现象二
- 在前
12个预测的LGC至RCP阶段区间,观察到两条斜率不同的直线,表明RCP阶段中,两种不同类型的反应中心(RCs)有不同的反应模式,即C-N键是否断裂(进行合成还是分解)。
- 在前
- 解释
RetroExplainer理解到,合成模式有利于降低能量分数;相应地,在LGC至RCP区间内的直线斜率为负。- 上述内容解释了去除苯环中氢原子的困难反应排在第二位的原因。
- 与排名第一、更容易合成目标产物的反应相比,苯环中移除的氢原子导致的能量差异较明显,表明决策曲线与反应困难程度之间存关联。
- 现象一
- 更多
RetroExplainer性能示例:见补充信息笔记5。
5.分子组装过程的子结构(substructure)归因能力
- 子结构归因能力
- 通过针对各阶段对最终能量得分的贡献比例的分析,分子组装过程能够在子结构(
substructure)层级实现归因能力。
- 通过针对各阶段对最终能量得分的贡献比例的分析,分子组装过程能够在子结构(
- 子结构归因能力的必要性
- 在涉及多个反应中心(
RCs)的情况下,多个化学键的数量变化,或者多个氢原子的变化,往往会合并到同一阶段,从而造成某种程度的不确定性。(图3c展示了六个有代表性的相关实例)
- 在涉及多个反应中心(
- 归因分析一——能量得分与反应难易程度的关联
- 现象:通过对比排名第一、第二的预测(图
3c中分子1、分子2),可以发现能量得分可能与反应的难易程度(例如选择性)相关。 - 原因:尽管
I:33与C:26之间的连接不利于能量的减少,但在C:26处添加一个氢原子所导致的能量增长约为前者的13倍(分别为13.5和1.02)。 - 以往研究的结论:
C–N交叉偶联反应通常涉及特定催化剂,并面临选择性问题。 I:33减弱了这种选择性问题,正与分子1的预测能量分数小于分子2的事实相对应。
- 现象:通过对比排名第一、第二的预测(图
- 归因分析二——正确答案能量得分被高估的原因
- 比较分子
3、4和5可得,正确答案的能量得分被高估的原因是,模型倾向于断裂C:26和N:27之间的化学键。 - 这一做法既能带来
5.40的能量收益(源自化学键断裂),又能因在N:27处增加一个氢原子而导致能量得分降低9.95。这两种形式的能量降低完全抵消了C:26处氢原子数增加所带来的能量增加影响。 - 因此,来自分子3的正确答案排名在分子
2和分子5的预测之后,因为它未能从C:26和N:27化学键断裂带来的能量减少中受益。
- 比较分子
- 归因分析三——空间位阻对模型推理的影响
- 现象:分子
4、分子6的分子结构相同,但预测出的能量分数不同。二者的唯一差别是,离去基团(LG)连接到了一对对称但编号不同的氮原子上。分子6的能量分数排名为21位。 - 原因
- 尽管这导致了在分子
6的预测中N:27和N:30处氢原子数量的变化,但它们相互抵消,对能量值总效应的影响仅为-0.53%。 - 相比之下,最重要的因素是由
C:33−N:30和C:33−N:27形成的化学键所引起的能量变化;而前者导致的能量变化几乎只有后者的十分之一。 - 这是因为后者的连接发生在化学键断裂之前,而
C:30与N:27之间的连接相对于N:30而言,会造成更大的空间位阻,最终导致能量增加。
- 尽管这导致了在分子
- 现象:分子
6.RetroExplainer对其他方法预测结果的重新排序
- 通过采用如图
3a所示基于能量的过程,RetroExplainer还可以对其他现有方法作出的预测进行重新排序。 - 重新排序算法伪代码:见补充信息笔记
6。 - 实验设定
- 选取
3种不同的逆合成方法(RetroXpert, GLN, NeuralSym)生成的预测结果,使用RetroExplainer进行重新排序。 - 选择原因:预测结果相对容易获取。
- 预测结果选取:对于每个目标产物,挑选前
50个预测结果进行重新排序;针对许多目标产物的预测结果不足50个的问题,引入一种基于平均百分比排名的评估策略。
- 选取
- 结果
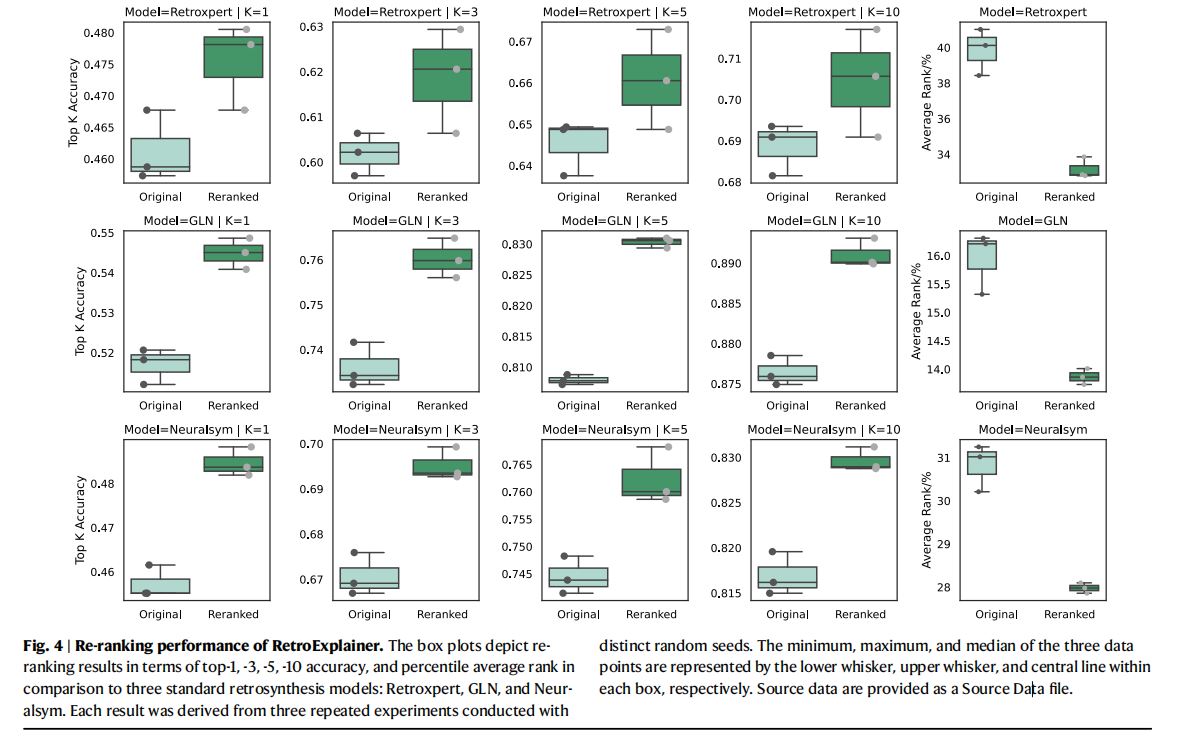
- 重新排序后的
top-1, top-3, top-5, top-10准确率结果如图4所示。
- 重新排序后的
- 实验结论
- 重新排序后,预测准确率取得显著提高。
RetroExplainer具有强大的重新排序能力,能够改善其他已有方法的预测结果。
三、RetroExplainer应用于多步逆合成预测
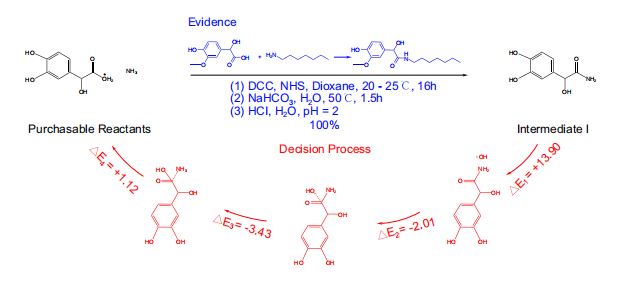
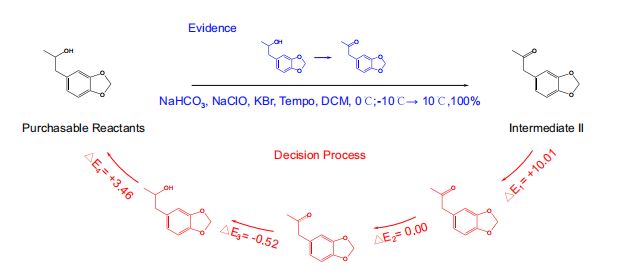
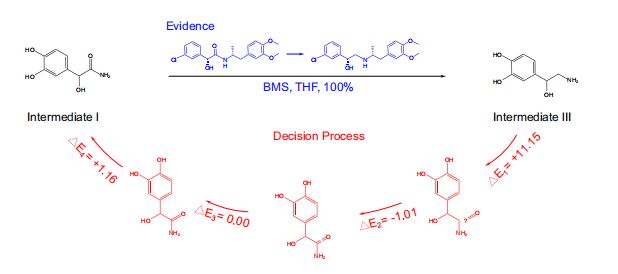
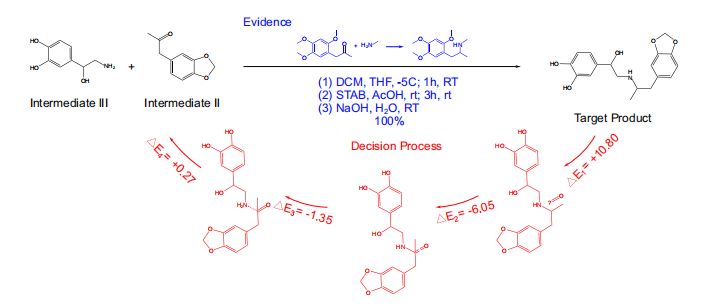

- 将RetroExplainer模型与Retro*算法、可购买分子列表相结合,用RetroExplainer替换了Retro的单步模型。
- 以protokylol(一种用于支气管扩张剂的β-肾上腺素受体激动剂)为例,说明RetroExplainer提供的解释。(图5)
- RetroExplainer为protokylol设计了一条四步合成路线。
- 决策过程的能量得分揭示了支持RetroExplainer做出相应预测的关键子过程。
- 方案的实用性
- 许多提出的反应未能找到文献中的直接对应结果,但能发现具有高产率,且与
RetroExplainer提出反应相匹配的类似反应。
- 许多提出的反应未能找到文献中的直接对应结果,但能发现具有高产率,且与
- 提供了包含
176个单步的101个路径规划案例,其中153个单步预测可以通过SciFindern引擎搜索40找到相似反应模式。 - 实验设置、多路径规划结果及文献检索发现的更多详细信息:见补充信息附录7、补充数据1和补充数据2。
四、反应类型对实验的影响
- 反应类型信息的注入方式
- 给出反应类型时,向
super node中添加额外嵌入。 - 经\(L\)个消息汇聚层后,
super node被提取为图级别的表征。
- 给出反应类型时,向
-
(表
1)引入反应类型后top-k准确率的提升
-
反应类型影响RetroExplainer性能的机制探究
- 根据隐藏特征来源(最后一个
RCP层/最后一个LGM层)、是否了解反应类型,提取4种类型的隐藏特征。 - (图6)反应类型对隐藏特征的影响
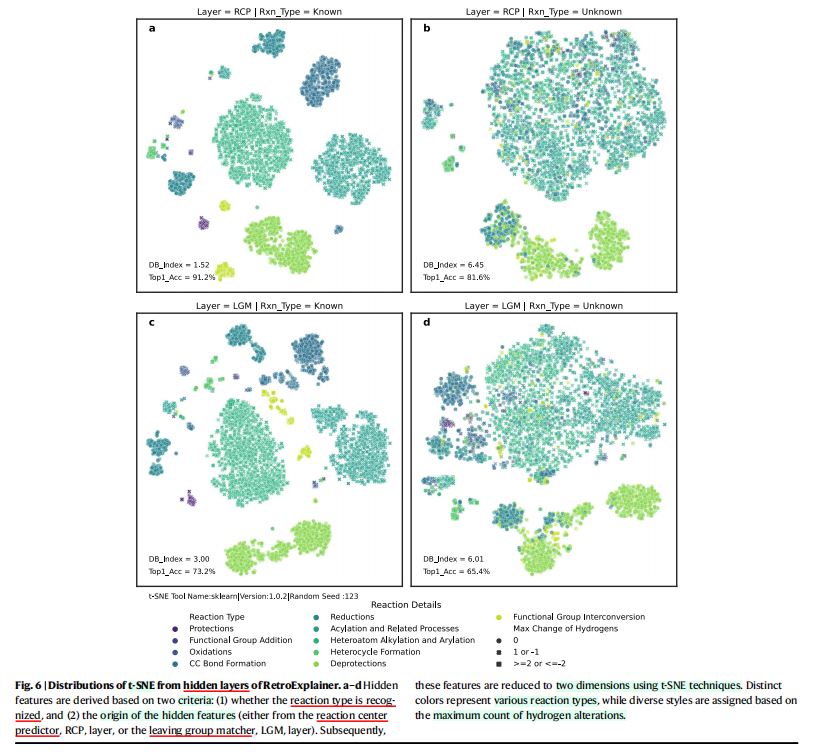
- 方法:使用
t-SNE(t分布随机邻居嵌入)对反应类型标签进行着色,以展示压缩隐藏特征的分布情况。 - 结论
- 相比于不受反应类型限制的情况,反应类型对产物隐藏表征施加了更为规范的约束。
- 约束对反应中心(
RCs)的定位过程、离去基团(LGs)的匹配过程进行增强,从而提高了整体性能。
- 根据隐藏特征来源(最后一个
Discussion——RetroExplainer局限性与未来研究展望
- 模型对离去基团(
LG)预测能力的局限性- 预测罕见
LGs性能受限LGM模块是基于预先收集的LGs数据库的预测分类器,简化了从补充信息附录3中的方程(S3.5)生成LGs的困难,并通过强先验保证了LGs的合法性(即所有LGs均来自预收集的数据库)。- 受到训练集质量的根本限制,
LGM方法在生成稀有LGs方面的灵活性有限,这是许多数据驱动模型共同面临的挑战。 - 即使是基于翻译的模型,在预测常见
LGs以便生成未曾见过的LGs方面也不够健壮。
- 模型对常见
LG的偏差- 尽管模型采用了如
SACL等策略,使得LGM模块能够在重新排序外源反应物模式期间,从数据库中找到与未见LGs相似的LGs作为代理(参考补充信息附录6中的算法),并扩大LGs数据库(在USPTO-FULL数据集中,作者收集了约7万个LGs),但仍无法完全消除这种偏向于常见LGs的偏差。 - 案例展示:见补充信息附录
9。
- 尽管模型采用了如
- 有前景的改进技术
- 对
LGM进行预训练 - 元学习
- 主动学习
- 数据增强
- 上述技术有望改善模型在处理罕见化学结构片段(即
LGs)预测问题时的性能和适应性。
- 对
- 预测罕见
- 决策过程灵活性受限
- 本实验涉及的分子组装过程
- 作者在逆合成预测中提出了一种类似于
SN2分子组装的过程,以生成带有能量的决策行动。 - 上述过程较为固定,虽然确实有效,但一些决策行动并不直观地符合其他不同的反应类型。
- 例如:没有明确
LGs(离去基团)参与的特殊反应类型,可能直觉上更适合SN2机制(案例:见补充信息附录10)。 - 对于不遵循
SN2机制的其他多种反应类型,当前方法可能不够灵活。
- 作者在逆合成预测中提出了一种类似于
- 可能的改进方法
- 引入其他反应机制
- 引入环加成反应、消去-加成反应等机制,反应机制可根据实际情况灵活选取,以设计更加灵活的决策过程。
- 增加反应机制选择模块
- 建议增加一个由
LGM(离去基团预测模块)和RCP(反应中心预测)置信度决定的机制选择模块,以判断哪种类型的机制更适合用来产生更易于人类理解的解释。
- 建议增加一个由
- 改进效果
- 模型能更好地适应各种不同类型的化学反应,并且生成的人工智能解释将更为贴近化学家的思维习惯,以及实际反应机制。
- 引入其他反应机制
- 本实验涉及的分子组装过程
- 无法进行细粒度预测
- 无法预测的详细反应信息
- 反应操作
- 温度
- 反应时间
- 反应条件预测已成为自动化合成平台发展过程中愈发紧迫的挑战,未来研究有待探索。
- 无法预测的详细反应信息
Methods
一、问题定义
1.基于图的逆合成预测
- 化学反应的一般表示
- \((\{G_{p;i}\}_{i = 1}^{N_p}, \{G_{r;j}\}_{j = 1}^{N_r})\),其中,\(G_p\)表示产物图,\(G_{r}\)表示反应物图。
- 反应推理模型
- \(f_{\theta}(\{G_{p;i}\}_{i = 1}^{N_p}, \{G_{r;j}\}_{j = 1}^{N_r})\),其中\(\theta\)是一组可学习的参数。
- 前向推理模型
- \(f_{\gamma}(\{G_{p;i}\}_{i = 1}^{N_p}, \{G_{r;j}\}_{j = 1}^{N_r})\)
- 逆合成模型
- 只有主产物输入,作为先验条件忽略了其余副产品。
- \(f_{\gamma}(\{G_{p;i}\}_{i = 1}^{N_p}, \{G_{r;j}\}_{j = 1}^{N_r}/\{G_{p:m}\}|\{G_{p:m}\})\),其中\(\{G_{p:m}\}\)表示主产物。
- 事实上,逆合成模型比前向模型更加复杂,前向模型的表现总体好于逆合成模型。
2.反应中心(Reaction Center, RC)和离去集团(Leaving Group, LG)
- 反应中心
- 子图\(G_c = \{(u, v), e_{uv}|e_{uv} \in G_{p}, e_{uv} \notin G_{r:j}, \forall G_{r:j} \in \{G_{r:j}\}_{i = 1}^{N_r}\}\),其中\((u, v)\)表示原子对,\(e_{uv}\)表示原子对之间的边。
- 合成子
- 一组图,根据反应中心,对产物图进行变换而成。
- 合成子通常并不在化学上有效,后续过程通过在合成子上添加离去集团(
LGs),形成最终反应物。
- 离去集团
- 反应物的子图,包含了并未出现于主产物之中的反应物原子和及其关联边。
3.基于GNN的Transformer
- 基于
GNN的Transformer的数据流- 一个句子,可以视为带有语义边(
semantic edge)的全连接图。 - 单词(
word tokens)作为结点进行处理。
- 一个句子,可以视为带有语义边(
- 多头注意力(
MHA)- \(m_a^{(l)}:= \sum_{w' \in N_{w}}\tilde{a}_{w'w}S_{w'}^{(l - 1)}W_v, m_{w}^{(l)}:= \tilde{a}_{ww}S_{w}^{(l - 1)}W_V \cdots (1)\)
- \(\tilde{a}_{w'w} = SOFTMAX(\frac{S_{w'}^{(l - 1)}W_Q(S_{w}^{(l - 1)}W_K)^T}{\sqrt{d_{k}}})\cdots (2)\)
- \(S_{w}^{''(l)} = COMBINE(m_a^{(l)}, m_{w}^{(l)}) := CONCAT(\{m_a^{(l:k)} + m_{w}^{(l:k)}\}_{k = 1}^{n_{head}})W^O\cdots (3)\)
- 上式中:\(N_{w} = S/{w}\)是结点\(w\)的语义邻域集合;\(d_{k}\)是\(W_k\)的隐藏维度数目。
RetroExplainer对基于GNN的标准Transformer的改进- 标准
Transformer无法处理拓扑空间中的边。 RetroExplainer中的Transformer解决了上述问题。
- 标准
二、RetroExplainer框架
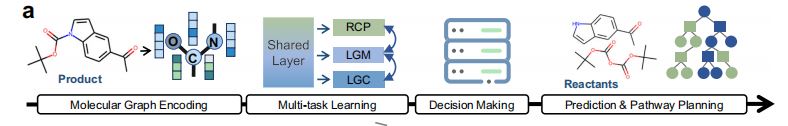
1.模型架构列举
(1)MSMS-GT, Multi-Sence and Multi-Scale Graph Transformer组件
(2)DAMT学习组件
(3)可解释的决策组件
(4)预测与路径规划组件
2.MSMS-GT, Multi-Sence and Multi-Scale Graph Transformer(多感知、多尺度图Transformer)组件
MSMS-GT模块(图1b右侧)
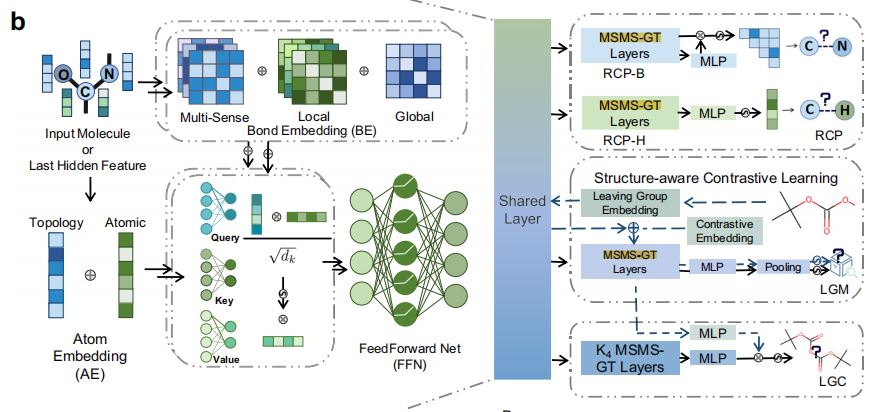
- 模型利用多感知、多尺度化学键嵌入策略,以及原子拓扑编码策略,以捕捉化学上重要的信息。
- 前两个编码器产生的分子向量通过多头注意力(
MHA)机制进行混合。
3.DAMT学习模块(图1b左侧)
- 产出的隐藏分子表示同时传送给三个特定任务头:RCP(Reaction Center Prediction)、LGM(Leaving Group Matching)和LGC(Leaving Group Completion)。
4.动态适应多任务学习(DAMT, Dynamic Adaptive Multi-Task Learning)技术(图1c)
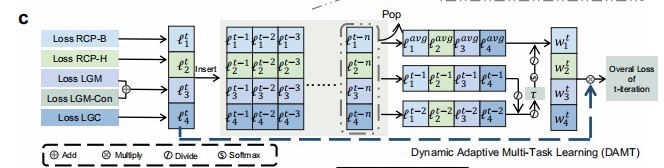
- 目的:平等训练每个子任务。
- RCP, Reaction Center Prediction:识别化学键和各个原子连接的氢原子个数变化情况。
- LGM, Leaving Group Matching:对于产物、产品离去基团数据库中的离去基团进行匹配。
- LGC, Leaving Group Connection:对离去基团和产物片段进行连接。
5.决策组件(图1d)
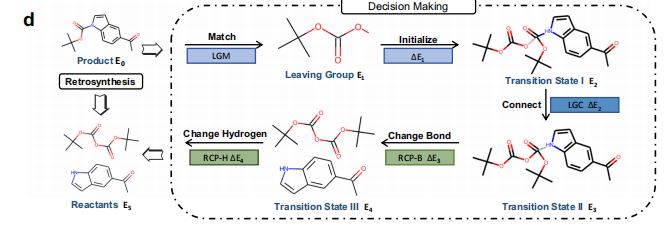
- 使用由5个逆合成动作,以及决策曲线能量分数组成的决策过程,将产物变换为反应物。
6.启发式树搜索算法
- 树搜索算法基于单步逆合成预测,融入最后一个组件,以发现高效的合成路线,使用透明的决策流程,同时保证反应物的可获得性。
三、多感知多尺度图Transformer(MSMS-GT)组件的细节
1.原子嵌入表示与拓扑嵌入表示(Atomic and Topological Embedding)
- 给定反应数据的
SMILES表示\(S_r\),产物的相应分子图\(G_{p} = (V, A)\)得到构建,其中原子被视为一个规模为\(N\)的结点集合。 - 将原子视为单词(
word tokens),构建语义域(semantic domain)的全连接图。 - 拓扑嵌入表示的引入
- 额外添加拓扑嵌入表示,目的:衡量原子在图的空间域中的重要性。
- 用拓扑嵌入表示替代传统位置编码,以免破坏最初的排列不变性。
- 使用结点的度数,用以描述原子的空间重要性。
- 初始化结点特征
- \(h_{\nu}^{(0)} = \sum_{i = 1}^{K_a}\phi_{x_i}(x_i) + \phi_d(deg(\nu))\)
- 带有不同下标的\(\phi(\cdot)\):分别表示不同的嵌入函数。
- \(x_{i}\):第\(i\)个原子特征(例如:原子序数、形式电荷等)。
- \(deg(\nu) \in \mathbb{R}\):表示原子\(\nu\)的总度数。
- 引入总度数的作用:作为强大的先验知识,使得注意力分数可以同时捕捉语义和空间信息。
2.多感知、多尺度化学键嵌入表示
- 引入化学键信息的作用:提高图的表达能力。
- 化学键性质编码
- 对于\(\sigma/\pi\)轨道、共轭键等化学键性质进行编码,编码后使用邻接矩阵\(\{A_{i} \in \{0, 1\}^{N \times N}\}_{i = 1}^{K_b}\)记录。其中,\(K_b\)是不同的感知。
- 不使用基于类型的方法进行边特征嵌入表示。
- 原因:直接揭示隐藏在化学键类型背后的反应相关属性(如:是否在环中),能提高模型对复杂反应类型的理解能力。
- 不同的化学键类习惯可能共享相同属性。
C-C, C=C均具有\(\sigma\)轨道。- 传统化学键类型编码无法表达这一信号。
- 化学键信息融入图数据
- 受到公式\((2)\)的启发(对基于原子对的语义边进行学习),作者考虑将化学键信息作为分子结构的一种先验。
- 将化学键信息添加到\(SOFTMAX(\cdot)\)之前的语义分数。
- 化学键特征嵌入公式
- \(m_{a}^{(l)} = \sum_{u \in N_{v}}\tilde{a}_{uv}h_u^{(l - 1)}W_V, m_{V}^{(l)} = \tilde{a}_{vv}h_{v}^{(l - 1)}W_V \cdots (5)\)
- \(a_{uv} = \frac{h_{u}^{(l - 1)}W_Q(h_v^{(l - 1)}W_k)^T}{\sqrt{d_k}} + \sum_{i}^{K_b}\phi_i(A_{i; uv}), \tilde{a}_{uv} = SOFTMAX(a_{uv}) \cdots (6)\)
- 上式中,\(N_{v} = V / \{v\}\)是原子\(v\)的邻域集合。
3.空间特征的嵌入表示
- 上述模型架构的表达能力仍然不如简单
GNN,简单GNN过分关注空间域中的1跳邻居。 Graphormer的解决方案- 使用一种最短路径嵌入表示。
- 全局距离\(A_g\)的计算
- 路径距离:由化学键长度进行计算
- 三维距离:由
MMFF优化得到的构象进行计算
- 引入全局距离嵌入表示后的
Transformer公式- \(a_{uv} = \frac{h_u^{(l - 1)}W_Q(h_v^{(l - 1)}W_K)^T}{\sqrt{d}_{k}} + \sum_{i}^{K_b}\phi_i(A_{i; uv}) + RBF(A_{g;uv}) \cdots (7)\)
- 高斯径向基函数:\(RBF(A) = \frac{exp(-(A - \phi_{mean})^2)/2\phi_{std}^2}{\sqrt{2\pi} \phi_{std}} \cdots (8)\)
- 全局距离的特点
- 引入空间信息,但总体上忽略了化学键信息。
- 原子环境(
AE, Atom Environment)- 将数个原子视为一个
token。 - 对于逆合成预测发挥了重要作用。
- 将数个原子视为一个
- 最终的注意力分数公式
- \(bias_{uv} = \sum_{i}^{K_b}\sum_{j = 1}^{K_r} \phi_i(A_{i; uv}^j) + RBF(A_{g;uv}) \cdots (9.1)\),第一项为局部项,第二项为全局项。
- \(a_{uv} = \frac{h_u^{(l - 1)}W_Q(h_v^{(l - 1)}W_K)^T}{\sqrt{d_k}} + bias_{uv} \cdots (9.2)\)
- \(A_i^j\):矩阵\(A_i\)的\(j\)次幂矩阵,描述了半径为\(j\)的原子环境。
- \(K_r\):原子环境的最大半径。
- 局部项:\(\phi_i(A_{i; uv}^j)\)
- 全局项:\(RBF(A_{g;uv})\)
- \(A_{i}^j, A_i \in\{0, 1\}^{N \times N}\):记录了各原子之间距离为\(j\)的路径数目,从而对半径为\(j\)的原子环境进行了描述。
- 最终注意力公式(公式\((9)\))的理解
- 利用自注意力机制,学习结点特征之间的语义关联性。
- 引入多层级环境\(\{A_i^j\}_{j = 1}^{K_r}\),以及多感知嵌入表示\(\{A_{i}\}_{i = 1}^{K_b}\),以在局部尺度充分利用丰富的化学键信息。
- 添加原子的全局嵌入表示,以在空间域捕捉全局范围。
- 结论:
RetroExplainer的表达能力至少与GNN等量齐观;此外,全局项\(RBF(A_{g;uv})\)允许了三维距离嵌入表示的引入(本实验结果:引入三维距离嵌入表示不是必要的)。
四、特定任务头(Specific Task Heads)、决策组件
1.特定任务头
- 设计
3个不同的特定任务头以满足不同需求。 RCP(Reaction Center Prediction):在判定反应中心时,针对各个化学键的变化给出一个概率分布。LGM(Leaving Group Matching):在产物和各个候选离去基团间,计算相容性分数。